SCINA on the cloud
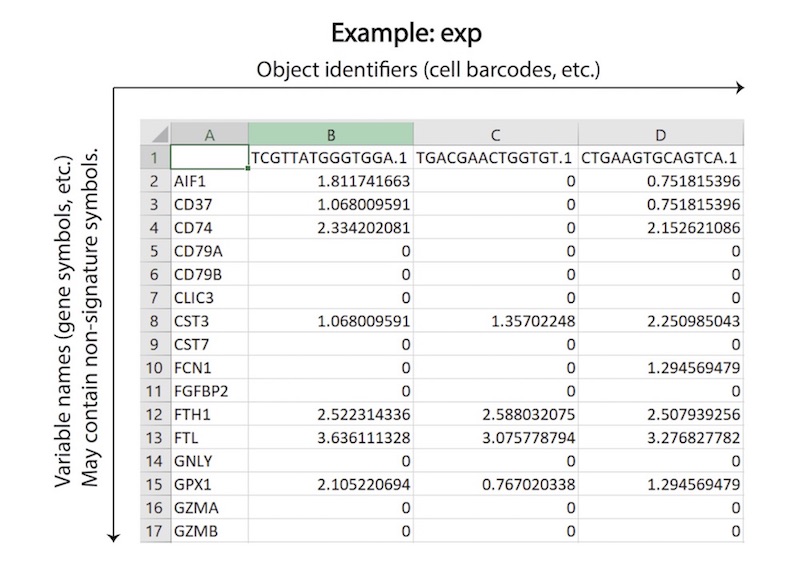
SCINA is regarded as semi-supervised, as the prior knowledge of signature genes is built into the unsupervised estimation process. SCINA accepts a list of signature gene sets for a variety of cell types, and an expression matrix, which is assumed to have been pre-processed by the user with logarithm transformation and/or any appropriate method of normalization if needed. For each cell type, the signature can have one or more genes. The signature genes, by default, should be highly expressed in one particular cell type compared to all other cell types. Expression of genes that are characteristically lowly expressed in one cell type compared to the other cell types can be inverted so that the pseudo expression of this gene is high in that cell type. Overlap in gene signature between cell types is allowed but should be kept to a minimum. After SCINA analyzation, the group of cells without high expression of any of the signature genes will be designated as an “unknown” class of cells (default mode), and the group of cells with high or low expression level of signature genes will be assigned a ‘cell label’ denoted by the names of the signature gene lists. SCINA also implements a switch that turns off the searching of “unknown” cells. Please find more technical details of SCINA from the manual of SCINA R package.